This example is taken from the ProBound web server, and corresponds to Figure 6 in the original Nature Biotech publication.
This example produces a model of the sequence-specificity of the tyrosine kinase c-Src by training on data from a Kinase-seq experiment, in which a bacterial display peptide library is exposed to c-Src and anti-phosphotyrosine pulldowns are performed at varying timepoints, which are modeled jointly.
import torch
import scipy.stats
import matplotlib.pyplot as plt
import pyprobound
import pyprobound.plotting
import pyprobound.fitting
Data specification
alphabet = pyprobound.alphabets.Protein()
url = "http://pbdemo.x3dna.org/files/example_data/Kinase/"
dataframes = [
pyprobound.get_dataframe(f"{url}countTable.0.200205_Src-Kinase_5m.tsv.gz"),
pyprobound.get_dataframe(
f"{url}countTable.1.200205_Src-Kinase_20m.tsv.gz"
),
pyprobound.get_dataframe(
f"{url}countTable.2.200205_Src-Kinase_60m.tsv.gz"
),
]
count_tables = [
pyprobound.CountTable(
dataframe,
alphabet,
left_flank="TAGTSVAGQSGQ",
right_flank="GGQSGQSGDYNK",
left_flank_length=3,
right_flank_length=3,
)
for dataframe in dataframes
]
Model specification
PSAMs
nonspecific = pyprobound.layers.NonSpecific(alphabet=alphabet, name="NS")
psam = pyprobound.layers.PSAM(
kernel_size=7,
alphabet=alphabet,
pairwise_distance=6,
seed=["***Y***"],
seed_scale=45,
symmetry=list(range(1, 8)),
frozen_parameters=pyprobound.layers.PSAM.frozen_positions(
[4], symmetry=list(range(1, 8)), out_channels=1, in_channels=20
),
name="Src",
)
Modes
modes = [
pyprobound.Mode.from_nonspecific(nonspecific, count_tables[0]),
pyprobound.Mode.from_psam(psam, count_tables[0]),
]
Rounds
round_0_5m = pyprobound.rounds.InitialRound()
round_1_5m = pyprobound.rounds.ExponentialRound.from_binding(
modes, round_0_5m, delta=-15, train_delta=False, target_concentration=0.25
)
round_0_20m = pyprobound.rounds.InitialRound()
round_1_20m = pyprobound.rounds.ExponentialRound.from_round(
round_1_5m, round_0_20m, target_concentration=1.0
)
round_0_60m = pyprobound.rounds.InitialRound()
round_1_60m = pyprobound.rounds.ExponentialRound.from_round(
round_1_5m, round_0_60m, target_concentration=3.0
)
Experiments
experiments = [
pyprobound.Experiment(
[round_0_5m, round_1_5m],
name="Src-5m",
counts_per_round=count_tables[0].counts_per_round,
),
pyprobound.Experiment(
[round_0_20m, round_1_20m],
name="Src-20m",
counts_per_round=count_tables[1].counts_per_round,
),
pyprobound.Experiment(
[round_0_60m, round_1_60m],
name="Src-60m",
counts_per_round=count_tables[2].counts_per_round,
),
]
Model
model = pyprobound.MultiExperimentLoss(experiments, pseudocount=50)
Fitting
optimizer = pyprobound.Optimizer(
model,
count_tables,
greedy_threshold=2e-4,
device="cpu",
checkpoint="Src.pt",
output="Src.txt",
)
optimizer.train_sequential()
tensor(2.3807)
optimizer.reload()
{'time': 'Tue Apr 23 19:42:08 2024',
'version': '1.3.1',
'flank_lengths': ((3, 3), (3, 3), (3, 3))}
Loss
with torch.inference_mode():
loss, reg = model(count_tables)
print(loss, reg, loss + reg)
tensor(0.5172) tensor(1.8635) tensor(2.3807)
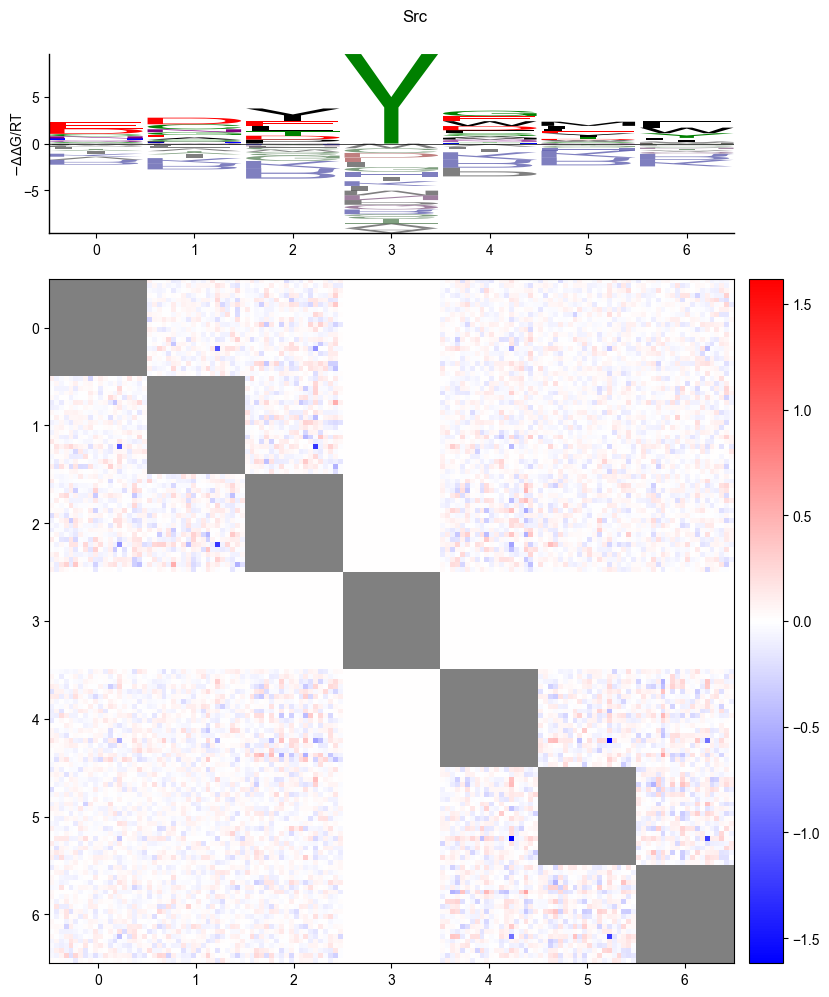
Logo
pyprobound.plotting.logo(psam)
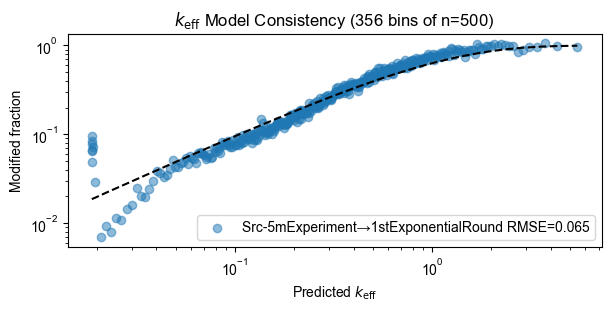
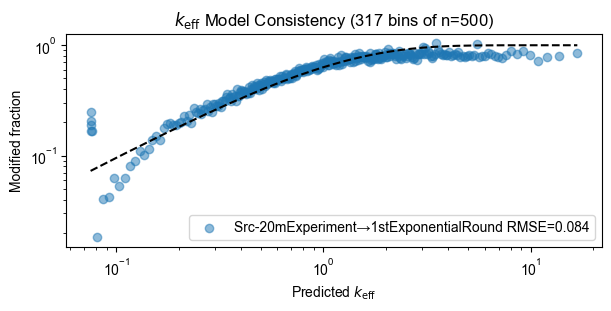
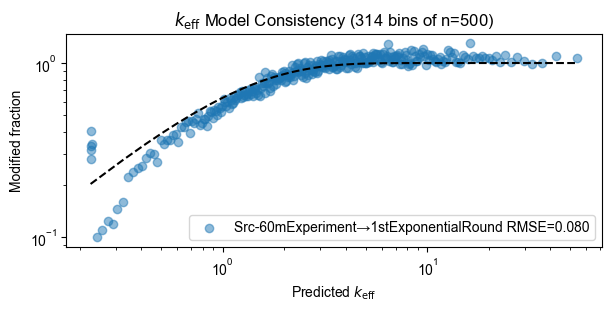
Model consistency
for experiment, count_table in zip(experiments, count_tables):
pyprobound.plotting.keff_consistency(experiment, count_table)
Validation
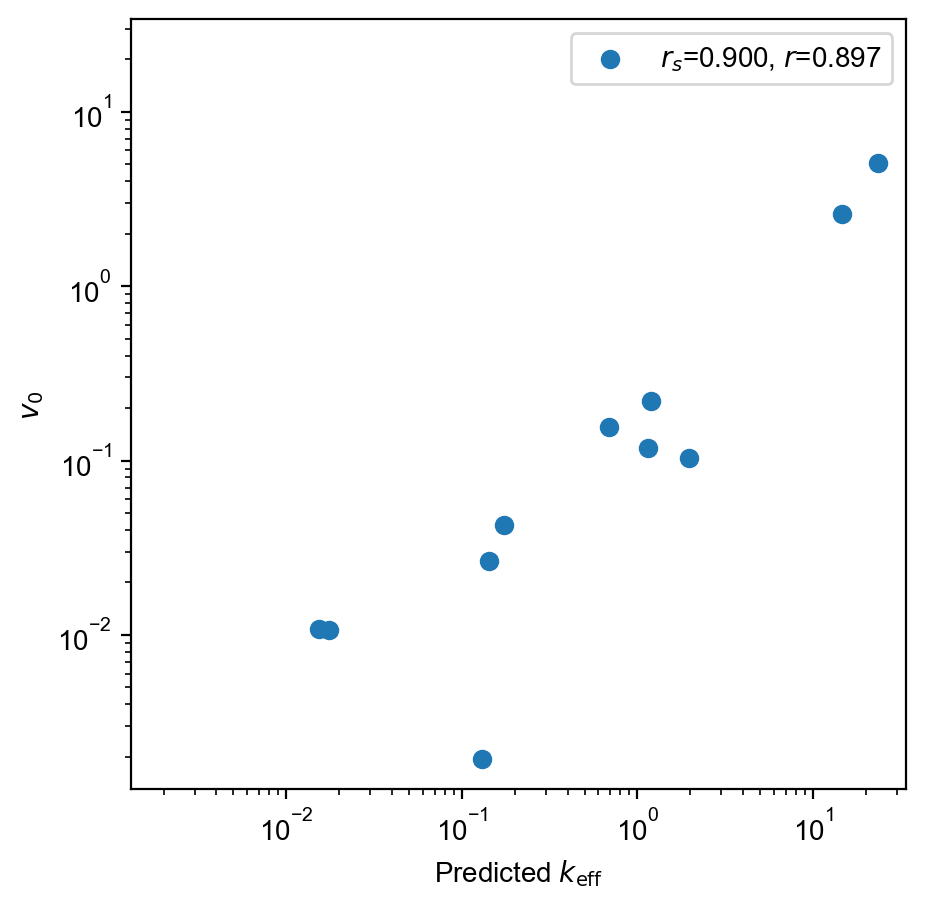
HPLC
validation_df = pyprobound.get_dataframe(["Src_HPLC.tsv"])
validation_ct = pyprobound.CountTable(validation_df, alphabet)
# Can manually score and plot sequences
observed = validation_ct.target[:, 0]
with torch.inference_mode():
predicted = torch.exp(round_1_5m.log_aggregate(validation_ct.seqs))
spearman_r = scipy.stats.spearmanr(observed.log(), predicted.log()).statistic
pearson_r = scipy.stats.pearsonr(observed.log(), predicted.log()).statistic
plt.figure(figsize=(5, 5))
plt.scatter(
predicted, observed, label=f"$r_s$={spearman_r:.3f}, $r$={pearson_r:.3f}"
)
plt.xscale("log")
plt.yscale("log")
plt.xlim(min(plt.xlim()[0], plt.ylim()[0]), max(plt.xlim()[1], plt.ylim()[1]))
plt.ylim(plt.xlim())
plt.xlabel(r"Predicted $k_{\mathrm{eff}}$")
plt.ylabel(r"$v_0$")
plt.legend()
plt.show()
# Or use built-in validation module with error bars
fit = pyprobound.fitting.LogFit(
round_1_5m,
validation_ct,
prediction=lambda log_aggregate: log_aggregate,
device="cpu",
update_construct=True,
name="Src HPLC",
)
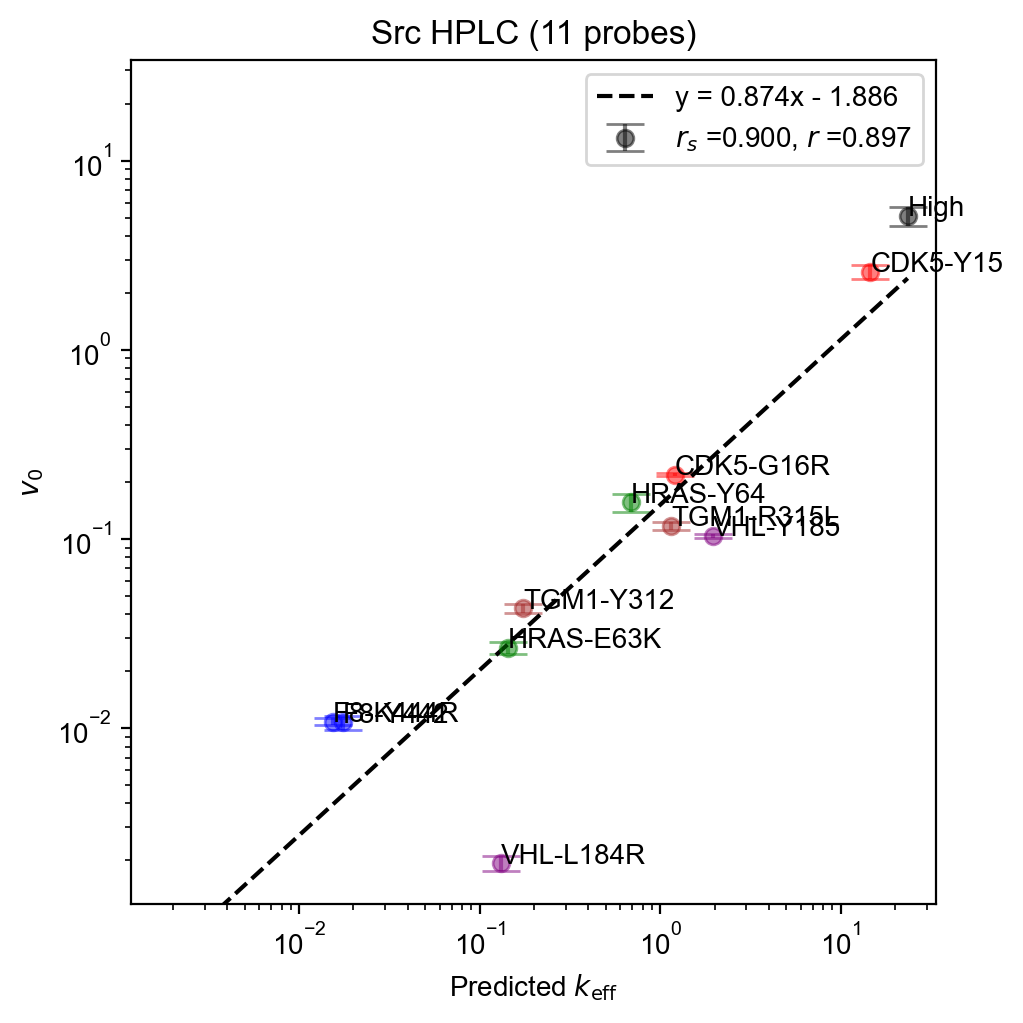
fit.plot(
xlabel=r"Predicted $k_{\mathrm{eff}}$",
ylabel=r"$v_0$",
labels=[
"High",
"F8-Y442",
"F8-K444R",
"TGM1-Y312",
"TGM1-R315L",
"VHL-Y185",
"VHL-L184R",
"HRAS-Y64",
"HRAS-E63K",
"CDK5-Y15",
"CDK5-G16R",
],
colors=(
["black"]
+ ["blue"] * 2
+ ["brown"] * 2
+ ["purple"] * 2
+ ["green"] * 2
+ ["red"] * 2
),
)