This example is taken from the ProBound web server, and corresponds to Figure 4 in the original Nature Biotech publication.
This example produces a single Dll binding model by training on Kd-seq data, which is an extension of a SELEX assay that sequences both the bound and unbound fractions, allowing for the calculation of absolute Kd’s from sequencing data.
import math
import pandas as pd
import torch
import scipy.stats
import matplotlib.pyplot as plt
import torch.nn.functional as F
import pyprobound
import pyprobound.plotting
import pyprobound.fitting
Data specification
alphabet = pyprobound.alphabets.DNA()
dataframe = pyprobound.get_dataframe(
"http://pbdemo.x3dna.org/files/example_data/"
"KD-single/countTable.0.20201205_DlldN-12.tsv.gz"
)
dataframe.head()
| 1 | 2 | 3 | |
|---|---|---|---|
| 0 | |||
| AAAAAAAAAA | 2 | 0 | 5 |
| AAAAAAAAAC | 3 | 0 | 3 |
| AAAAAAAAAG | 3 | 0 | 0 |
| AAAAAAAAAT | 2 | 0 | 4 |
| AAAAAAAACA | 2 | 0 | 5 |
count_table = pyprobound.CountTable(
dataframe,
alphabet,
left_flank="GAGTTCTACAGTCCGACCTGG",
right_flank="CCAGGACTCGGACCTGGA",
left_flank_length=6,
right_flank_length=6,
)
Model specification
PSAMs
nonspecific = pyprobound.layers.NonSpecific(alphabet=alphabet, name="NS")
psam = pyprobound.layers.PSAM(
kernel_size=10,
alphabet=alphabet,
pairwise_distance=9,
seed=["--TAATTG--"],
seed_scale=6,
name="Dll",
)
Modes
modes = [
pyprobound.Mode.from_nonspecific(nonspecific, count_table),
pyprobound.Mode.from_psam(psam, count_table),
]
Rounds
initial_round = pyprobound.rounds.InitialRound()
bound_round = pyprobound.rounds.BoundRound.from_binding(
modes, initial_round, target_concentration=100, library_concentration=20
)
unbound_round = pyprobound.rounds.UnboundRound.from_round(bound_round)
Experiment
experiment = pyprobound.Experiment(
[initial_round, bound_round, unbound_round],
name="Dll",
counts_per_round=count_table.counts_per_round,
)
Model
model = pyprobound.MultiExperimentLoss([experiment], pseudocount=200)
Fitting
optimizer = pyprobound.Optimizer(
model,
[count_table],
greedy_threshold=2e-4,
device="cpu",
checkpoint="Dll.pt",
output="Dll.txt",
)
optimizer.train_sequential()
tensor(0.9410)
optimizer.reload()
{'time': 'Wed Apr 24 02:43:16 2024',
'version': '1.3.1',
'flank_lengths': ((6, 6),)}
Loss
with torch.inference_mode():
loss, reg = model([count_table])
print(loss, reg, loss + reg)
tensor(0.8029) tensor(0.1381) tensor(0.9410)
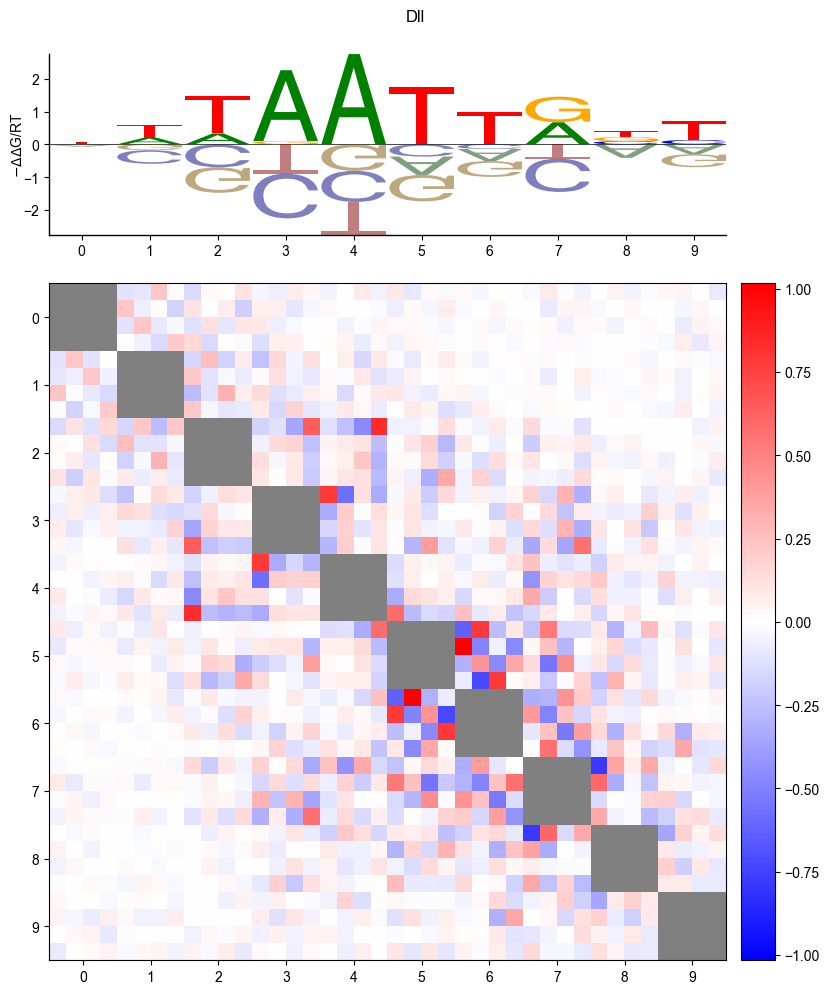
Logo
pyprobound.plotting.logo(psam)
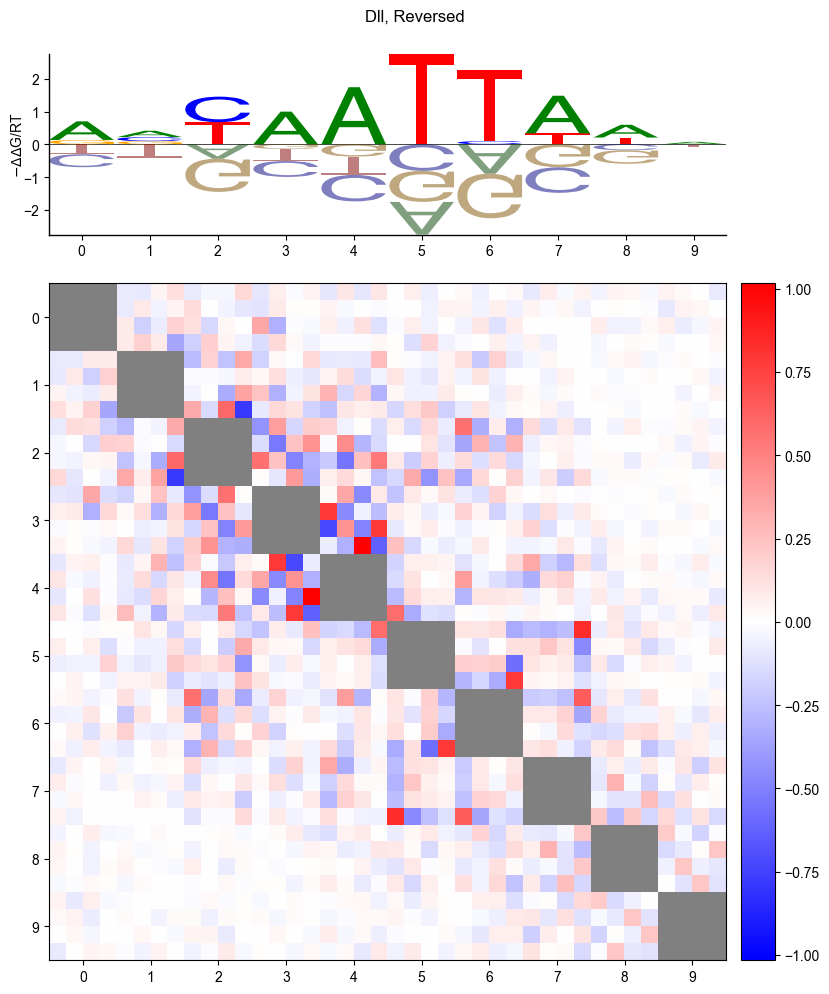
pyprobound.plotting.logo(psam, reverse=True)
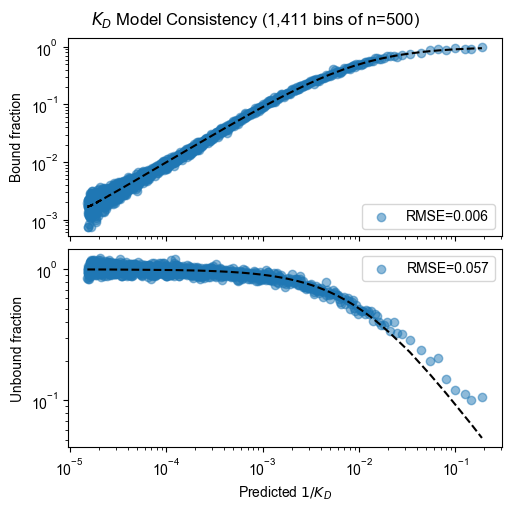
Model consistency
pyprobound.plotting.kd_consistency(experiment, 0, 1, 2, count_table)
Validation
EMSA
validation_df = pyprobound.get_dataframe(["Dll_EMSA.tsv"])
validation_ct = pyprobound.CountTable(
validation_df,
alphabet,
left_flank=count_table.left_flank,
right_flank=count_table.right_flank,
left_flank_length=count_table.left_flank_length,
right_flank_length=count_table.right_flank_length,
)
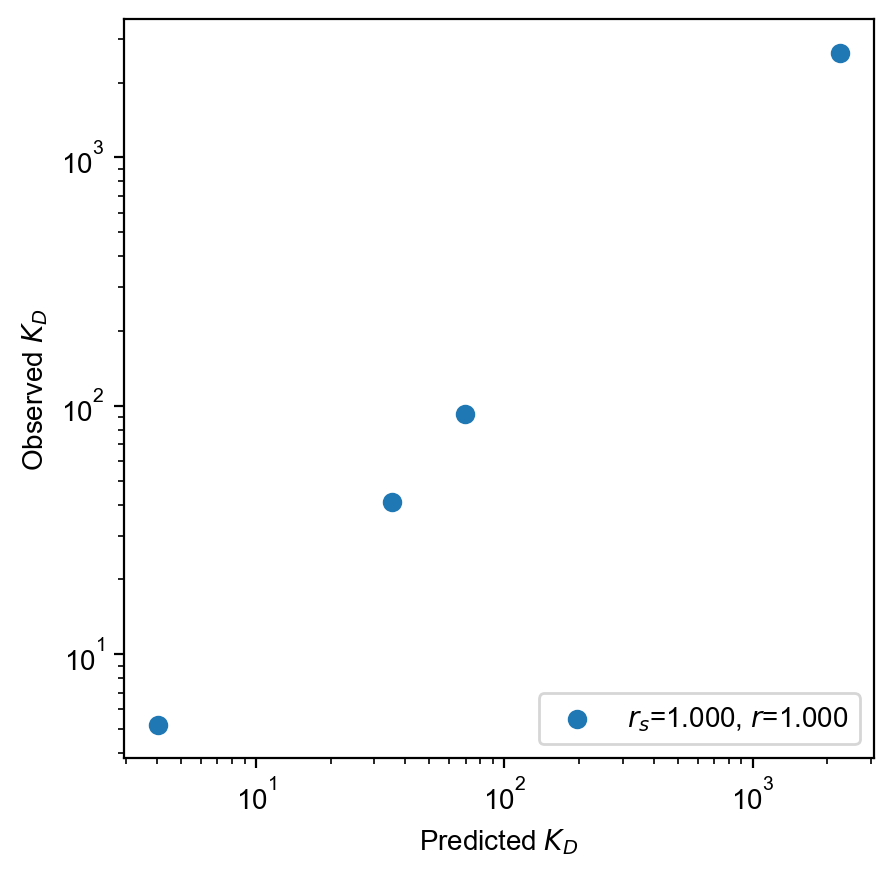
val_free_protein = experiment.free_protein(0, 1, 2, library_concentration=6.7)
# Can manually score and plot sequences
observed = validation_ct.target.squeeze()
with torch.inference_mode():
predicted = val_free_protein / torch.exp(
bound_round.log_aggregate(validation_ct.seqs)
)
spearman_r = scipy.stats.spearmanr(observed.log(), predicted.log()).statistic
pearson_r = scipy.stats.pearsonr(observed.log(), predicted.log()).statistic
plt.scatter(
predicted, observed, label=f"$r_s$={spearman_r:.3f}, $r$={pearson_r:.3f}"
)
plt.xscale("log")
plt.yscale("log")
plt.axis("scaled")
plt.xlabel(r"Predicted $K_D$")
plt.ylabel(r"Observed $K_D$")
plt.legend(loc="lower right")
plt.show()
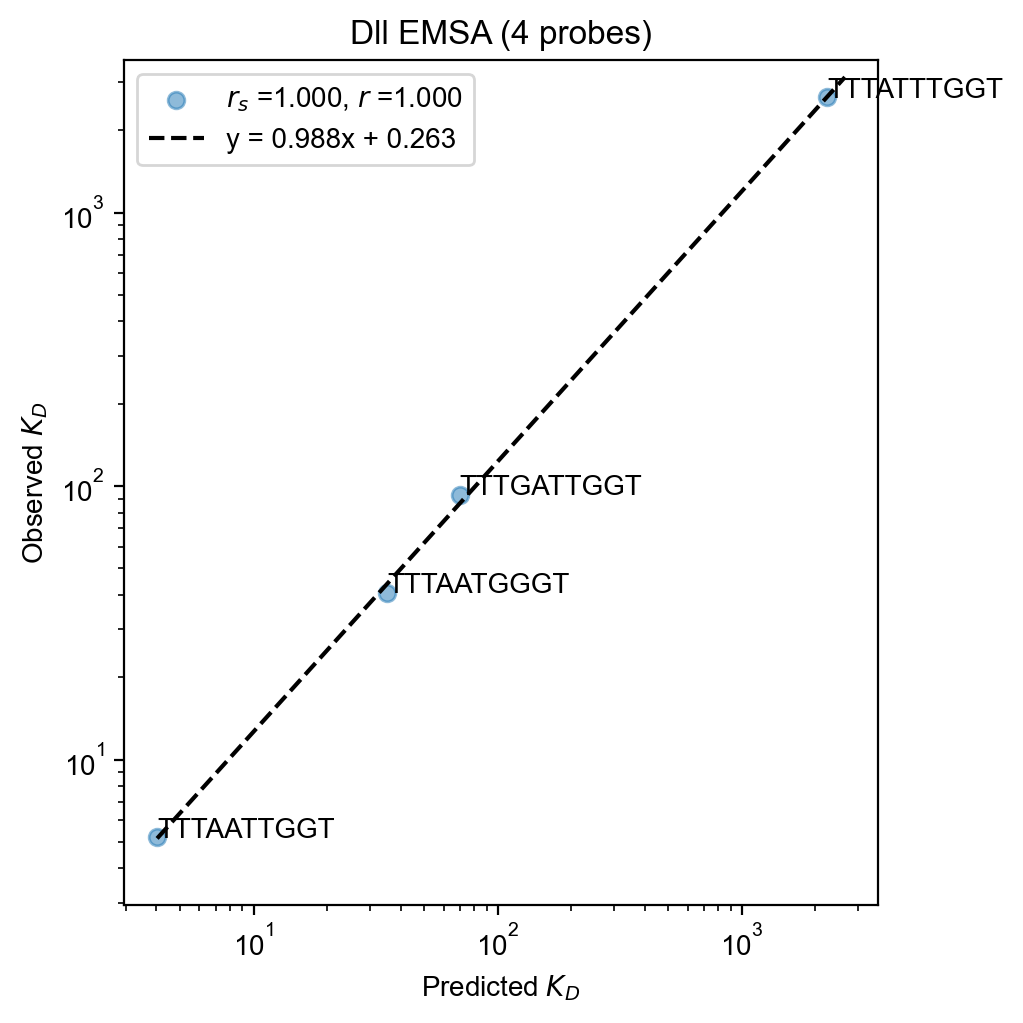
# Or use built-in validation module
prediction = lambda log_aggregate: math.log(val_free_protein) - log_aggregate
fit = pyprobound.fitting.LogFit(
bound_round,
validation_ct,
prediction,
device="cpu",
update_construct=False,
name="Dll EMSA",
)
fit.plot(
labels=validation_df.index,
xlabel=r"Predicted $K_D$",
ylabel=r"Observed $K_D$",
)
PBM
# PBM table generated from Weirauch et al. (2014)
pTH5506_HK_df = pd.read_csv(
(
"https://www.ncbi.nlm.nih.gov/geo/download/"
"?acc=GSM1291486&format=file&file=GSM1291486"
r"%5FpTH5506%5FHK%5F8mer%5F2086%2Eraw%2Etxt%2Egz"
),
header=0,
index_col=None,
sep="\t",
compression="gzip",
)
pTH5506_HK_df.index = (
pTH5506_HK_df["linker_sequence"] + pTH5506_HK_df["pbm_sequence"]
)
pTH5506_HK_df = pTH5506_HK_df.loc[pTH5506_HK_df["control"] == "FALSE"]
pTH5506_HK_df = (
pTH5506_HK_df["mean_signal_intensity"]
- pTH5506_HK_df["mean_background_intensity"]
).to_frame()
pTH5506_HK_ct = pyprobound.CountTable(
pTH5506_HK_df, alphabet, right_flank="-" * 9, right_flank_length=9
)
pTH5506_HK_df.head()
| 0 | |
|---|---|
| CCTGTGTGAAATTGTTATCCGCTCTGCCAGTTTAGGTGGCGCCCGGAACCCTTAACCCAT | 1335.8308 |
| CCTGTGTGAAATTGTTATCCGCTCTCATGTAGAGCCCTAAAACTGGGACTAAGCCGACCT | 1369.9194 |
| CCTGTGTGAAATTGTTATCCGCTCTGGACGCAACATGCAGCTGCACAAGTCACTTGTGAG | 2527.1627 |
| CCTGTGTGAAATTGTTATCCGCTCTAAGATTGACACGAGACTATCCAGTATACCCCTTTC | 1905.6660 |
| CCTGTGTGAAATTGTTATCCGCTCTGTGCTCGAAGAAAGGGCCACCGCGTCCCTCGCTAG | 1392.8326 |
fit = pyprobound.fitting.LogFit(
bound_round,
pTH5506_HK_ct,
prediction=F.logsigmoid,
update_construct=True,
train_offset=True,
train_posbias=True,
train_hill=True,
device="cpu",
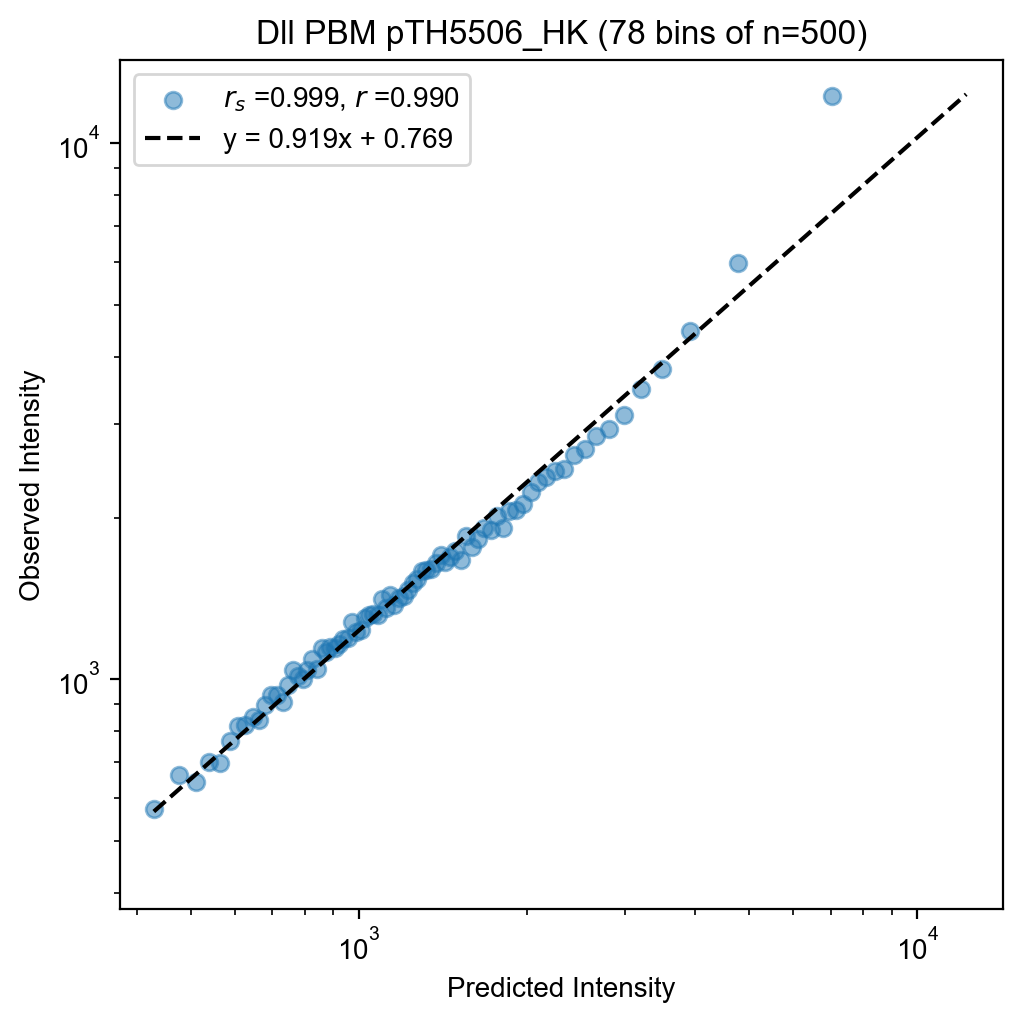
name="Dll PBM pTH5506_HK",
checkpoint="Dll_pTH5506_HK.pt",
)
fit.fit()
fit.plot(kernel=500, xlabel="Predicted Intensity", ylabel="Observed Intensity")